In this paper, we present an enhancement to Apache Spark, which is a distributed computing framework, to accommodate efficiently SQL queries on genomic datasets. Although one can use existing technologies to import genomic datasets as SQL tables, the poor performance of those technologies to common genomic queries has made them an unattractive choice among users of Next Generation Sequencing (NGS) data.
As sequencing costs drop, more and more research centers invest in massive sequencing projects that aim to build huge databases of thousands of genomes and their associated phenotypic traits. For example the Oregon Health and Sciences University (OHSU) and the Multiple Myeloma Foundation (MMRF) are sequencing 1000 patients with Acute Myeloid Leukemia (AML) and Multiple Myeloma respectively [1, 2]. In another example, the International Cancer Genome Consortium (ICGC) [3] is in the process of sequencing over 25,000 pairs of tumor and normal samples in order to catalogue the genetic abnormalities of 50 different cancer types. Each whole genome sequencing run with Illumina’s technologies produces more than 200 GB of data.
Access to those data, though crucial for the advancement of cancer treatment, remains a challenge for researchers and data scientists.
Today there are two tiers of data access: a top and bottom tier. The top tier involves the downloading of FASTQ, BAM, or VCF files from an archive such as the SRA [4] or CGHUB [5] that contain reads or variants from the sequencing either of a person or a population. Although those archives use the state of the art of file sharing technology to reduce file transfer latencies over the Internet - as is the case of CGHUB that uses GeneTorrent [6] - the size of the files that need to be transfered makes downloading slow. For example, the downloading of a 250 GB BAM with a 60x coverage of Whole Genome Sequencing (WGS) over a 100 Mbps Internet link takes 8 h. On the other hand, the bottom tier involves extractions of subsets of the downloaded data. A user either develops software from scratch to navigate through the data, or they use a combination of shell scripts in combination with commands of either of vcftools, samtools, and BEDtools. This practice adds a layer of complexity between data and the user for three reasons:
1. Scripts must be created to analyze these experiments
2. It requires users to manually parallelize the execution of those tools in distributed environments which get adopted to serve the increasing amounts of generated data.
3. It creates storage overheads with the possible creation of intermediate files that are used to transform files.
Assuming that genomic data in the order of Terabytes and Petabytes reside in distributed environments as in [7], we propose that a more efficient alternative to both tiers of data access is a distributed data retrieval engine. Such an interface on the top access tier can provide data on demand by eliminating the network traffic and the need for secondary level processing at the user end. Even if owners of genomic data repositories are reluctant to provide such a feature, a data retrieval interface is still useful for the productivity of an end user at the bottom layer. With such an interface end users will not have to worry about scripting their way to retrieve and compare data from datasets of different origin, such as raw reads, variants, and annotation data.
In this work we use Spark SQL which is the distributed SQL execution engine of the Apache Spark [8] framework. Spark is a user friendly high performance framework; it abstracts distributed collections of objects and it provides around 80 operators that either transform those objects through opeators such as map, filter, and groupBy, or perform actions on them through operators such as reduce, count, and foreach. Spark organizes a cluster in a MasterSlave architecture, where the driver (i.e., the master) executes a main program and transfers code to workers (i.e. slaves) to execute on those parts of the distributed objects that they contain.
Data model
In this work we assume that all genomic data are in ADAM format. ADAM [9, 10] is an open source software that separates genomic information from its underlying representation and is currently used as the main computational platform in the NIH BD2K center for big data in translational genomics [7]. Such a separation removes from data users the burden of how data is represented. Thus, an ADAM user can operate on distributed storage without having to parse complicated files, given that ADAM supports and replaces all levels of genomic data that are currently represented by the legacy FASTQ, BAM, and VCF formats.
ADAM records consist of serializable objects that are stored in a distributed friendly column based format. It uses Apache AVRO [11] as a data serialization system, which relies on schemas and stores them together with the data. The serialized data are stored with the use of Apache Parquet [12] system, which is a columnar storage system based on Google Dremel [13]. Parquet creates storage blocks by grouping sequences of records and it stores sequentially all columns of each block. Finally, given that Parquet provides built in support for writes and reads from the Hadoop File System (HDFS), ADAM transparently supports distributed environments that are built over HDFS.
Spark SQL fully recognizes Parquet files and consequently ADAM files as relational tables and it also infers their schemas. This allows users to natively query ADAM files from SPARK SQL.
The problem
Although Spark SQL provides impressive expressive power and thus it can execute any genomic query, the main obstacle for its adoption to query genomic data has been its slow performance on two of the most frequently encountered queries: 1) random range selection, and 2) joins with interval keys. Random range selection in a collection of aligned reads took several minutes to run in a small cluster, which is embarrassingly slow given that samtools need only a few seconds. Fortunately, the rapid evolution of the open source libraries that we use (in particular Parquet on which ADAM files depend and whose API Spark SQL uses for their filtering) improved the execution of those queries by an order of magnitude as we show in the results section. Regarding the execution of interval joins between two tables, Spark SQL uses the obvious execution of filtering on their cross product. However, given the sizes that are involved in genomic data such an approach is unrealistic. If we consider for example an interval join between 1 billion aligned reads with 1 million variants, the cross product between them is 1015 records and it is prohibitively slow to compute.
The contribution of this paper addresses the second performance bottleneck: joins on interval keys. We present a modification to Spark SQL that enhances the efficiency of interval joins and it thus makes it suitable to query genomic data. For this reason we use interval trees to interval join two tables in a distributed setting.
Related work
The first generation of tools that access genomic data involves packages such as Samtools [14], vcftools [15], BAMtools [16] and BEDtools [17]. While powerful, these tools require extensive programming expertise to open and parse files of different formats, assign buffers, and manipulate various fields. In addition, given that these tools are optimised for single node computational performance, a user needs to manually parallelize them in a distributed environment.
The second generation of relevant software involves the Genome Query Language (GQL) [18], which provides a clean abstraction of genomic data collection through a SQL-like interface. However, the support of GQL is only limited to queries on a small subset of fields of the entire SAM specification and it also requires extra manual effort to support distributed environments.
The third generation leverages the Hadoop ecosystem to easily provide data on demand on a distributed setting. For example, the GenomeMetric Query Language (GMQL) [19] uses Apache Pig, which is a high level language that abstracts map-reduce operations, to support metadata management and queries between variants and annotation data. In another example, NEXTBIO [20] uses HBase, which is Hadoop’s NoSQL key-value store, to support data of similar nature. The scope of these tools however excludes raw data either in the FASTQ or in the BAM format.
Implementation
This section describes how we modified Spark SQL to add support for range based joins. The first step of the modification involves the grammar of Spark SQL, which we extended to simplify the syntax of those queries. Next, before describing our modification to the execution engine of Spark SQL, we provide a brief description of the interval tree and interval forest data structures which this modification utilizes.
Syntax
Although the existing syntax of Spark SQL suffices for a user to describe a join between two tables on interval overlap condition, it looks complex and counter intuitive for a user of genomic collections that routinely uses this operation. If we consider for example tables A (aStart: long, aEnd: long, aChr: String) and B (bStart: long, bEnd: long, bChr: String), then according to the current syntax of Spark SQL, an interval join looks like the following:
SELECT ∗ FROM A JOIN B ON ( aChr = bChr AND a S ta r t < aEnd AND b S ta r t < bEnd AND ( aStart < bStart AND aEnd > bStart OR bStart < aStart AND bEnd > aStart ) )
To eliminate the complexity of such a frequent operation, we enhanced the vocabulary of Spark SQL with two extra keywords, namely RANGEJOIN and GENOMEOVERLAP. In case of an interval based join, the former keyword replaces JOIN and the latter, which is followed by two tuples as arguments, specifies the overlap condition and is the only argument of the ON condition.
Using those new keywords, one can type the query of the previous example as follows:
SELECT ∗ FROM A RANGEJOIN B ON GENOMEOVERLAP( ( aS tar t , aEnd , aChr ) , ( bS ta r t , bEnd , bChr ) )
Interval trees
The most expensive part of a Join evaluation involves a search for overlaps between two arrays of intervals. Our implementation utilizes the interval tree data structure, which is a binary tree that is built from a collection of n intervals in O (n log n) time and it takes O (log n) time to find which of the intervals of its collection overlap with a given query interval. Note that a brute force execution, which is what Spark SQL uses up to date, of the same operation takes quadratic time.
At this point we remind interested readers how an interval tree is constructed and searched.
Each node of the tree contains two objects. The first is the key that is the mid point of the union of the collection of intervals that are stored in the subtree which is rooted at a node. The second object is an overlapping list that contains those intervals that overlap with the key. Consider for example the interval tree of Fig. 1 which stores
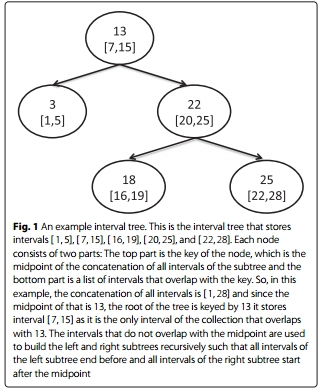
intervals [ 1, 5], [ 7, 15], [ 16, 19], [ 20, 25] and [ 22, 28]. The key of the root is 13 because that is the mid point of the union of all intervals which is [ 1, 28]. This key overlaps only with interval [ 7, 15], which is the only content of the overlapping list of the root.
The subtrees of a node store those intervals that do not overlap with its key. The left subtree contains all intervals the end points of which are less than the key; symmetrically, the right subtree contains all intervals with start points greater than the key.
To search whether a particular interval overlaps with any of the intervals of an interval tree, one scans linearly the overlapping list of the root to search for intervals that might overlap with the query and continues traversing the tree according to the relevant position of the query interval and the keys of the encountered nodes. When the input interval ends before the key of a node the search continues only to the left subtree. Respectively, when the start of the query interval is greater than the key the search continues only to the right. In case of overlapping between an input interval and a key, the search continues to both subtrees. Assume for example a search for all overlapping intervals with [ 17, 23] with the interval tree of Fig. 1. Starting from the root, after a quick scan of the overlapping list of the root returns the empty set, a comparison between the key of the root and the interval indicates that the search should continue towards the right subtree, which is rooted at the node with 22 as key. A quick scan of the overlapping list of the node detects that [ 20, 25] is part of the solution and since the query interval overlaps with the key of the node, the search continues towards both left and right subtrees. Proceeding in the same way, the query returns as a result the set of intervals [ 20, 25], [ 16, 19] and [ 22, 28].
